中国科大揭示半导体表面位点特异的化学键成键和非绝热传能机制
中国科学技术大学蒋彬教授课题组近期在半导体表面的非绝热动力学理论研究方面取得重要进展。研究成果以“Real-Time Coupled Electron-Nuclear Dynamics of Chemical Bond Formation on a Semiconductor Surface”为题,4月16日发表于国际著名学术期刊《美国化学会志》(Journal of the American Chemical Society)。
气体原子或分子在固体表面的散射与反应,是催化、能源转化以及半导体器件制造等过程中的核心微观机制。其中,化学键形成过程中涉及的电子与原子核耦合运动(非绝热动力学)长期以来缺乏清晰认识。相关研究较多的是金属表面,例如H原子与金属表面碰撞中绝大部分能量都转移到金属电子激发中,这也是气相H原子得以吸附在金属表面所必要的非绝热能量耗散过程。相关现象可以被简单的电子摩擦理论进行描述,其中H原子核运动时受到金属电子连续激发产生的平均阻尼,进而形成核到电子的非绝热能量耗散。
相比金属表面,半导体由于具有有限带隙,其电子激发与能量耗散机制更加复杂,使得相关问题更具挑战性。近年来,实验在Ge(111)-c(2×8)重构表面观测到氢原子散射过程中的显著非绝热效应:当入射能量超过表面带隙时,散射氢原子的能量损失呈现出明显的“双峰分布”,并表现出位点选择性特征。然而,传统基于Born-Oppenheimer近似的绝热动力学模拟以及基于电子摩擦理论的动力学模拟均无法对这一现象给出合理解释。
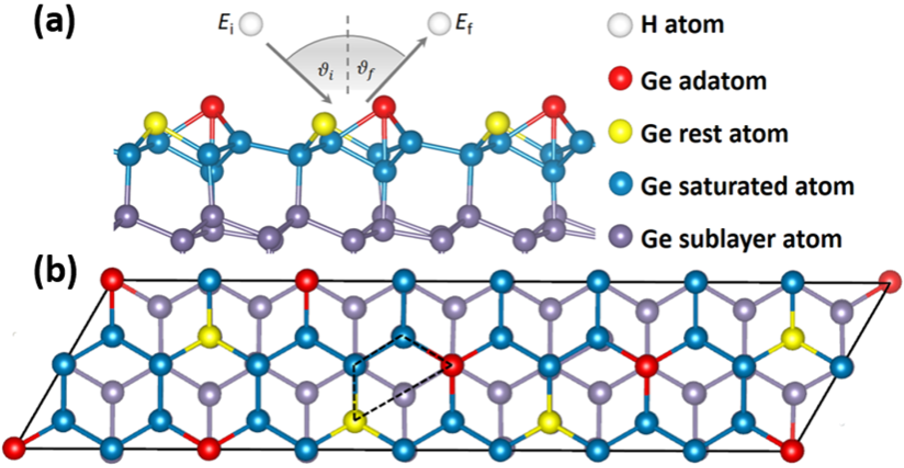
图1 Ge(111)c(2×8)重构表面结构
在此背景下,研究团队应用实时含时密度泛函理论(rt-TDDFT)与Ehrenfest动力学相结合的第一性原理方法,实现了对电子与原子核运动耦合过程的“实时”模拟。该方法能够在原子运动过程中同步描述电子结构演化,从而捕捉复杂的非绝热动力学行为。研究结果在氢原子平动能损失以及散射角分布等方面均与实验观测高度一致,成功再现了实验中发现的双峰能量损失特征。进一步分析表明,这一双峰结构来源于氢原子在重构表面不同Ge原子位点上的“位点选择性”碰撞行为:当氢原子与Ge表面的rest atom发生碰撞时,会形成瞬态Ge–H键,并触发超快的跨位点电子转移过程,从而产生显著的电子激发和较大的能量损失;而当氢原子与adatom相互作用时,尽管同样存在瞬态成键过程,但几乎不发生电子转移和电子激发,因此仅表现出较小的能量损失。这些结果表明,rt-TDDFT方法能够有效描述不同表面位点上化学键形成过程中显著不同的超快电子-核运动耦合动力学,为理解半导体表面非绝热能量传递机制提供了关键理论依据。
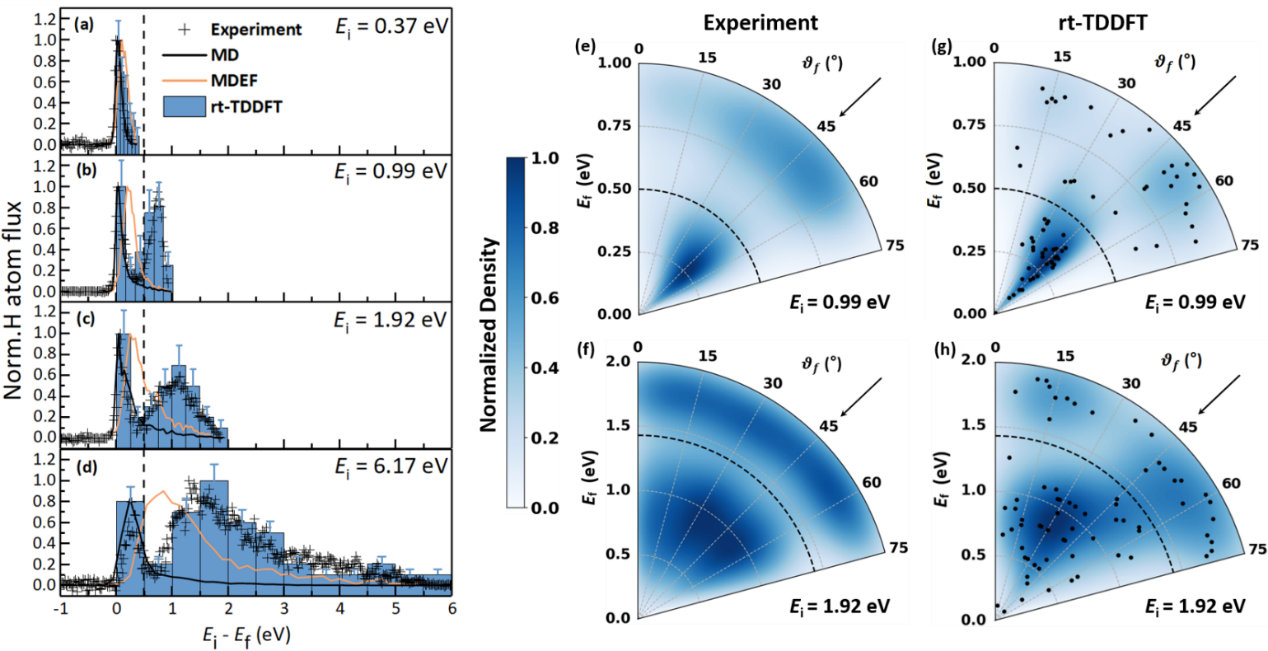
图2 理论模拟与实验平动能损失以及散射角分布对比。
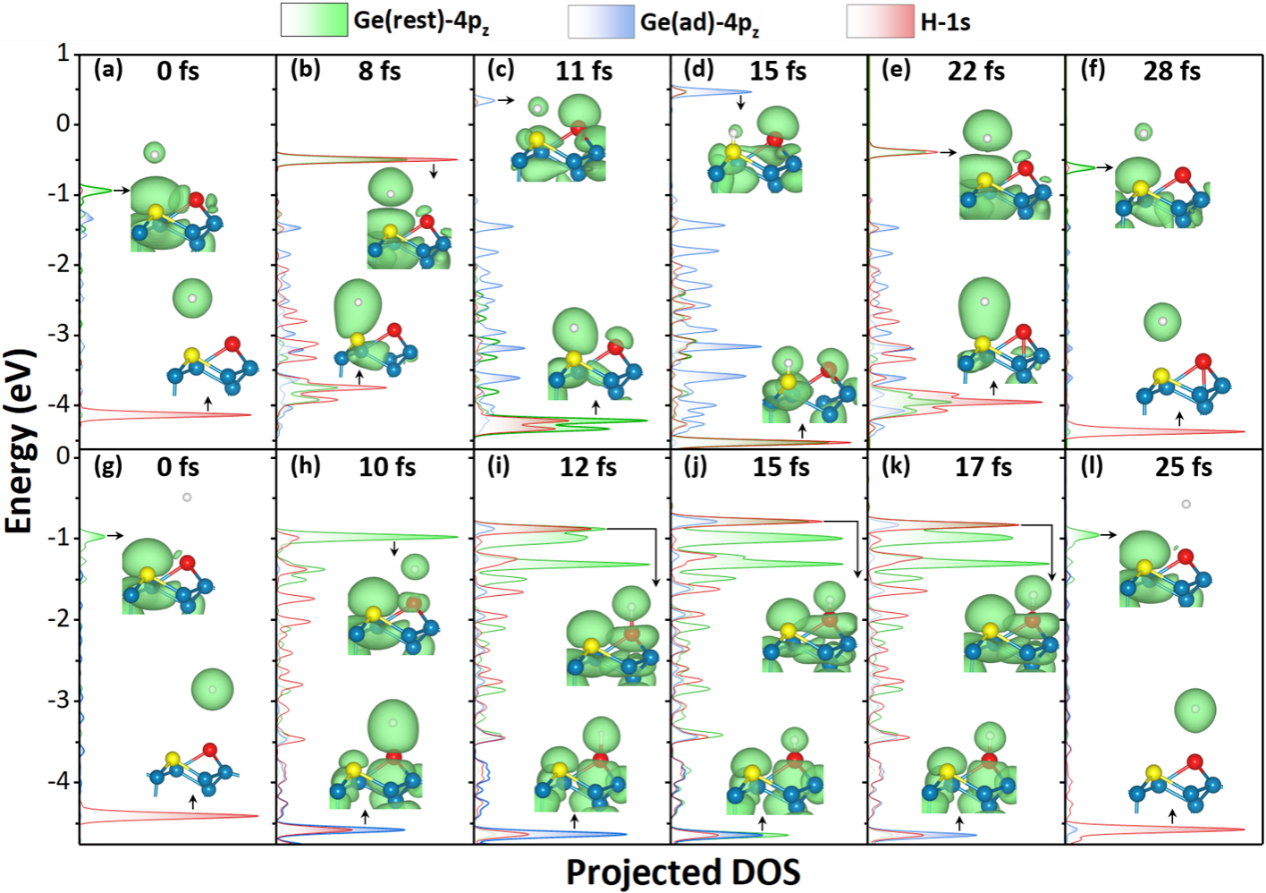
图3 投影态密度与轨道分辨电子密度的时间演化过程。
化学与材料科学学院、化学国家高层次人才培养中心博士生史佳龙为该论文的第一作者,蒋彬教授为通讯作者。该工作得到了量子科学与技术创新项目、基金委杰出青年基金等基金的资助。
论文链接:https://pubs.acs.org/doi/full/10.1021/jacs.5c22902
(化学与材料科学学院、精准智能化学全国重点实验室、科研部)




