近期,中国科大学蒋彬教授课题组在发展高精度机器学习方法上取得新进展,最新成果以“Physically Motivated Recursively Embedded Atom Neural Networks: Incorporating Local Completeness and Nonlocality”为题发表于《物理评论快报》(Physical Review Letters) 上。
原子模拟可以帮助我们在微观层面理解分子光谱、反应动力学和能量/电荷转移过程。发展精确且高效的势能面对于模拟这些过程至关重要。近年来,随着机器学习技术的发展,原子神经网络框架已经成为构建势函数的常用方法。在这个框架下,体系总能量可以拆分为每个原子能量之和,原子能量被认为是原子局部化学环境的函数,长期以来人们都认为基于三体的描述符已经足够描述局部的原子化学环境。但近期有工作发现这些基于三体甚至四体相互作用的描述符会导致一些非物理的原子能量简并,不能完备的描述局部化学环境。这会使得目前绝大多数原子型神经网络势能面的训练会受到这种简并扭曲构型空间的影响,难以进一步提高精度。
蒋彬教授课题组长期致力于发展高精度机器学习势能面方法,受量子化学中原子轨道组合为分子轨道的方式启发,研究团队改进了前期发展的嵌入原子神经网络方法(J. Phys. Chem. Lett. 2019, 10, 4962),使得嵌入电荷密度描述符中的轨道系数变为化学环境依赖,以递归的通过更新嵌入电荷密度描述符实现,提出递归嵌入原子神经网络方法(图1)。有趣的是,这种神经网络方式与物理上不太直观的消息传递神经网络形式本质上相同。研究团队进一步证明可以通过递归更新轨道系数的形式来引入更多体相互作用,推导出完备的描述一个局部化学环境,确定迭代次数(消息传递的次数)与近邻原子数之间的关系。该方法无需显式计算高阶相互作用,极大的简化了计算,并从多体相互作用的角度解释了消息传递型神经网络的优越性。通过在甲烷和体相水这些体系上数值测试结果显示,递归嵌入神经网络在准确性上明显优于现有机器学习模型(图2),验证了这个新模型的局部完备性和非局域性。该研究提出一个通用的策略可以很容易地改进现有的机器学习势能面框架以包括更复杂的多体相互作用描述符,而无需改变其原始的基本结构,这将为开发更准确、更高效的机器学习模型提供一个新的思路。
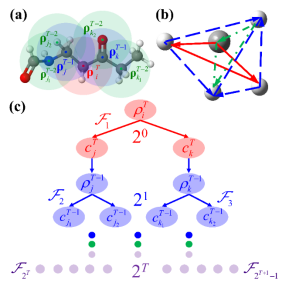
图1:(a)递归嵌入原子神经网络模型的示意图显示了密度描述符是如何递归嵌入的;(b)以CH4为例展示了如何通过两次迭代实现以C为中心的完备五体相互作用;(c)图示说明每次迭代三体项的数量如何增加。
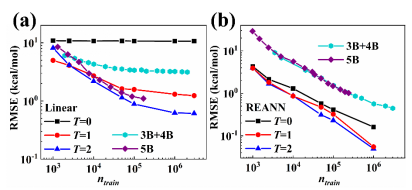
图2:递归嵌入原子神经网络与包含5体以及3体+4体的机器学习模型构建CH4分子势函数的精度比较,图 (a)和图 (b)分别是线性拟合和非线性拟合的结果。
张耀龙为该论文的第一作者,蒋彬教授为通讯作者。该工作得到了国家重点研发计划、国家自然科学基金委、安徽量子信息计划的资助。
相关论文链接:https://journals.aps.org/prl/pdf/10.1103/PhysRevLett.127.156002
(合肥微尺度物质科学研究中心、化学与材料科学学院、科研部)




