近日,中国科学技术大学合肥微尺度物质科学国家研究中心、化学与材料科学学院黄伟新教授和华东理工大学龚学庆教授、中国科学技术大学张群教授、中国科学院大连化学物理研究所范峰滔研究员合作研究了优先暴露不同晶面的锐钛矿TiO2纳米晶光催化甲醇氧化反应,揭示了TiO2表面位点敏感的界面电荷转移和光催化反应效率。研究成果于3月8日以“Site Sensitivity of Interfacial Charge Transfer and Photocatalytic Efficiency in Photocatalysis: Methanol Oxidation on Anatase TiO2 Nanocrystals”为题发表在Angew. Chem. Int. Ed.上。
光催化利用绿色可持续太阳能驱动化学反应,从而受到广泛关注。光催化包括光催化剂吸收光并生成载流子、载流子分离并扩散至光催化剂表面、载流子与光催化剂表面物种发生氧化还原反应等步骤。前两个步骤发生在光催化剂体相,通过优化光催化剂结构来提高其效率获得了巨大进展。第三个步骤发生在光催化剂表面,涉及到光生电荷在吸附物种-光催化剂界面的转移,是光催化反应的速控步,但由于其复杂性,相关研究较少。
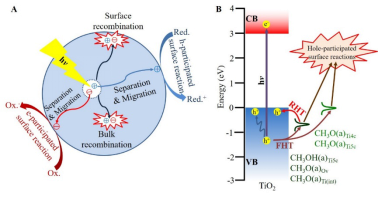
研究人员首先利用时间分辨原位漫反射红外光谱表征了TiO2纳米晶表面吸附甲醇和甲氧基物种光催化氧化反应动力学,观察到表面物种光催化氧化反应速率常数敏感依赖于表面位点。同一纳米晶表面,甲氧基物种光氧化反应性能优于甲醇;不同纳米晶表面,甲氧基物种光催化氧化反应性能随甲氧基/甲醇比例增加而提高。TiO2-{001}纳米晶表面表现最大的甲氧基:甲醇比例,因此其表面甲氧基表现出最高的光催化氧化反应活性,而表面甲醇为光催化氧化反应惰性。理论计算结果表明钛矿TiO2表面吸附甲醇物种及其能级结构与TiO2表面结构密切相关。在TiO2(001)表面主要生成4配位Ti吸附的CH3O(a)Ti4c物种和少量5配位Ti吸附的CH3OH(a)Ti5c物种;在TiO2(100)和(101)表面生成CH3OH(a)Ti5c、5配位Ti吸附的CH3O(a)Ti5c和少量与缺陷相关的甲氧基和甲醇物种。CH3O(a)Ti4c和CH3O(a)Ti5c的HOMO位于对应TiO2表面的价带顶,而其它物种的HOMO位于价带顶1eV以下。因此TiO2价带中的光生空穴更易转移至CH3O(a)Ti4c和CH3O(a)Ti5c物种并只能发生氧化反应,而转移至其它物种的光生空穴既可以发生氧化反应,也可以重新转移至TiO2价带。对应的瞬态吸收光谱结果证实甲醇-TiO2-{001}纳米晶界面仅发生TiO2纳米晶至甲醇的光生空穴转移过程,而甲醇-TiO2-{100}纳米晶和甲醇-TiO2-{101}纳米晶界面不仅发生TiO2纳米晶至甲醇的光生空穴转移过程,而且发生甲醇至TiO2纳米晶的光生空穴转移过程。同时,CH3O(a)物种增强TiO2表面能带向上弯曲,利于光生空穴向表面的扩散;而CH3OH(a)削弱TiO2表面能带向上弯曲,不利于光生空穴向表面的扩散。原位表面光电压谱结果证实具有最大的甲氧基/甲醇比例的甲醇/TiO2-{001}体系表现出最强的能带向上弯曲程度,因此其CH3O(a)Ti4c表现出最高的光催化氧化反应活性。
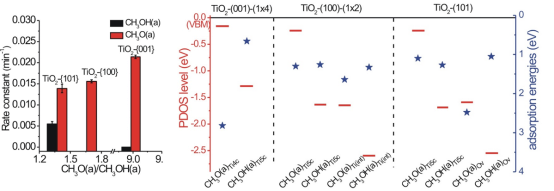
该研究结果展示了TiO2纳米晶光催化甲醇氧化反应中TiO2表面位点和甲醇吸附物种依赖的TiO2表面能带弯曲/体相至表面载流子扩散过程和电子结构/界面电荷转移过程,表明光催化剂表面结构优化是是实现光催化效率最大化的有效途径。值得指出的是,该甲醇/TiO2纳米晶体系光催化表面化学与黄伟新教授课题组之前的甲醇/TiO2单晶体系光催化表面化学(J. Am. Chem. Soc. 135 (2013) 5212; Angew. Chem. Int. Ed. 55 (2016) 623; J. Phys. Chem. 123 (2019) 31073/121 (2017) 9991)相一致,充分证实了黄伟新教授提出的“从单晶到纳米晶模型催化体系”概念的科学性和可行性(Acc. Chem. Res. 49 (2016) 520; Surf. Sci. Rep. 74 (2019) 100471)。
文章第一作者是中国科学技术大学博士研究生傅聪,共同第一作者是华东理工大学博士研究生李菲和中国科学技术大学博士研究生张佳晨。该项研究得到了国家自然科学基金委员会、科技部、中国科学院、教育部、安徽省等的支持。
论文链接:https://onlinelibrary.wiley.com/doi/epdf/10.1002/anie.202014037
(合肥微尺度物质科学国家研究中心、化学与材料科学学院、科研部)




