5月20日,中国科大信息科学与技术学院、生物医学工程研究中心李骜研究组以原创性论文(Original paper)的形式,在生物信息学领域国际权威刊物Bioinformatics上在线发表了最新研究成果(CLImAT: accurate detection of copy number alteration and loss of heterozygosity in impure and aneuploid tumor samples using whole-genome sequencing data)。该论文的第一作者是信息学院电子科学与技术系2013级博士研究生余振华,通讯作者为其研究生导师、信息学院副教授李骜,其他作者分别为刘元宁、沈艺和王明会。中国科大是论文的第一单位,也是唯一单位。
基因组异常是多种恶性肿瘤的标志性特征,在肿瘤发病机理、临床诊断和治疗等研究中具有极为重要的作用,但传统的肿瘤基因组异常检测技术存在着通量低、分辨率差等问题。随着近年来大规模平行测序实验技术的快速兴起,新一代测序(Next-generation sequencing)凭借其在通量和分辨率方面的独特优势,目前已经成为癌症基因组学研究中最流行的实验手段。但由于肿瘤本身的复杂性,从新一代测序数据中准确检测基因组异常仍面临着正常细胞掺杂和污染、肿瘤基因组非整倍体性等棘手问题。目前利用新一代测序技术检测肿瘤基因组异常的方法呈现井喷式的发展,但这些方法没能提供可靠且全面的基因组变异的信息。
李骜研究组近年来在传统的SNP-array平台用于检测肿瘤基因组异常方面积累了丰富的经验,但基于新一代测序技术的肿瘤相关的研究才刚刚起步。在接近一年半的时间里,李骜研究组已经开发出了一整套处理和分析下一代测序数据的软件和方法,有效地解决了肿瘤样本分析中涉及到的关键性问题。目前更深入的肿瘤相关的研究正在进一步的展开。
在该论文中,李骜研究组提出一种基于全局参数化隐Markov模型的新颖生物信息学算法,通过期望最大化(Expectation maximization)方法对模型进行训练和参数估计,从而有效解决了利用新一代测序技术检测复杂肿瘤全基因组异常的国际性难题。审稿人对该论文的创新性给予了高度评价,认为该算法具有令人关注的特性并为解决上述问题提供了极佳的思路。
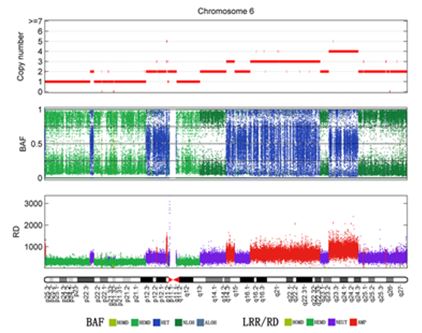
复杂肿瘤基因组异常的可视化检测结果
Bioinformatics杂志是生物信息学领域的权威学术刊物,其刊登的原创性论文注重介绍通过计算方法研究生物学问题中所取得的突破性发现,具有很高的学术认可度。2013年度中国大陆以第一单位发表在该杂志的原创性论文仅有17篇,占当年发表论文总数的2%。此前,中国科大为第一单位在Bioinformatics上发表的原创性论文不超过6篇。
李骜博士于2005年毕业于中国科大信息科学与技术学院,后赴美进行博士后研究。自2011年1月加入中国科大,担任信息科学与技术学院轨道制副教授、博士生导师,其研究组至今已在Bioinformatics、Nucleic Acids Research、Methods、PLOS ONE、IET Systems Biology、BMC Bioinformatics、Amino Acids、Molecular Biosystems等生物信息学领域知名刊物上发表SCI论文10余篇。
附论文链接:
http://bioinformatics.oxfordjournals.org/content/early/2014/05/19/bioinformatics.btu346.abstract
(信息学院)




